Ein klinisches Risikomanagement – heute im Krankenhaus-/Klinikalltag unverzichtbar – bedarf einer Strategie, die von der Leitung des Hauses definiert wird. Oberstes Ziel eines klinischen Risikomanagements ist es, die Patientensicherheit zu erhöhen und dadurch Patientenschäden zu vermeiden.
Den Vorgaben des Gesetzgebers folgend, haben viele Einrichtungen inzwischen damit begonnen, ein klinisches Risikomanagement einzuführen. Nun gilt es, die Maßnahmen berufsgruppenübergreifend und strukturiert umzusetzen, damit sie dem Patienten zugutekommen und Schäden oder unerwünschte Ereignisse vermieden werden. Was ist zu beachten? Welche Prozessschritte sind sinnvoll und notwendig?
In diesem Beitrag illustrieren wir anhand dreier Schadenfälle, die sich im Zusammenhang mit Implantationen von Hüftprothesen ereignet haben, klassische Haftungsrisiken, die nach unserer Erfahrung häufig zu Patientenschäden führen. Zudem erläutern wir, wie diesen Risiken mit festgelegten Präventionsmaßnahmen zielführend begegnet werden kann.
Klassische haftungsrechtliche Risiken bei der Implantation der Hüft-TEP
Die Implantation von Hüftprothesen gehört in Deutschland zu den Operationen, die sehr häufig vorgenommen werden. Praktisch jeder Haftpflichtversicherer, der im Krankenhausmarkt agiert, sieht sich irgendwann mit Schäden aus diesem Bereich konfrontiert, denn sie zählen zu den „Klassikern“ unter den Arzthaftpflichtschäden. Dass diese Schäden (für den Versicherer) mitunter sehr teuer werden können, zeigen die folgenden drei Beispielfälle.
Fall 1
Eine 40-jährige Patientin, die sich vor gut einem Jahr einer Hüftoperation nach dem Verfahren McMinn unterzogen hat, begibt sich mit der Diagnose Pfannenlockerung erneut ins Krankenhaus, um die damals eingesetzte Hüftprothese austauschen zu lassen. Bei der Zweit-OP wird die vorher implantierte Kappenprothese durch eine Kurzschaftprothese ersetzt.
Der Anwalt der Patientin behauptet (entgegen der Aussage des Krankenhauses), dass die postoperativen Röntgenbilder bereits nach der ersten OP keine einwandfreie Implantatlage gezeigt hätten. Vielmehr sei hier bereits ein deutliches Einsinken der Pfanne erkennbar. Dies ist laut Anwalt der wahre Grund für die Notwendigkeit der Revisions-OP (Entfernung der Schraubenpfanne über den ursprünglichen sowie einen neuen Zugang, Einsatz eines Spenderknochens und einer Pfannenabstützschale sowie Austausch des Keramikkopfs gegen einen Metallkopf).
Das Krankenhaus ist der Ansicht, dass der Sitz der Pfanne bei der Entlassung korrekt war, weist allerdings darauf hin, dass der tiefe Pfannenboden wegen eines bereits vorbestehenden Knochenschwundes keine Tragkraft mehr gehabt habe. Vor diesem Hintergrund sei die Patientin vor der Entlassung noch einmal eingehend befragt worden, ob Beschwerden bestünden, die evtl. auf einen Pfanneneinbruch hätten hindeuten können. Da die Patientin keine Beschwerden angegeben habe, hätten die Ärzte ihr zu diesem Zeitpunkt keinen erneuten Pfannenwechsel vorgeschlagen. Man habe, so die Begründung, zunächst abwarten wollen, ob es zu einer knöchernen Integration kommt.
Die verantwortlichen Ärzte vertreten den Standpunkt, dass das Auftreten der Pfannenlockerung mit Einsinken der Pfanne der schlechten Knochenqualität der Patientin geschuldet und somit den individuellen Gegebenheiten anzulasten sei. Die Notwendigkeit einer Revisions-OP sehen die Ärzte daher als schicksalshaft an.
Mit Verweis auf diese Argumentation lehnt der Haftpflichtversicherer des betroffenen Krankenhauses die Haftung ab, wogegen die Patientin über ihren Anwalt Klage einreicht.
Der vom zuständigen Landgericht beauftragte Gutachter kommt zu dem für das Krankenhaus negativen Ergebnis, dass die zuständigen Ärzte den intraoperativ bzw. direkt postoperativ evident gewordenen instabilen Zustand im Bereich der linken Hüfte nicht korrekt adressiert hätten. Der Befund der ins Becken hinein luxierten Hüfte sei unverständlicherweise bereits während des stationären Aufenthalts der Patientin negiert worden. Die Revision hätte nach Ansicht des Gutachters sofort erfolgen müssen. Daher lastet er den Ärzten einen groben Behandlungsfehler an – ein Ergebnis, das er einige Monate später in einem Ergänzungsgutachten noch einmal bekräftigt.
Als Reaktion auf dieses Ergebnis bietet der Haftpflichtversicherer der Patientin 400.000 € an für die Abgeltung des Schmerzensgeldes, des Verdienstausfalls, der Behandlungskosten und des Haushaltsführungsschadens.
Risikobewertung
In diesem Fall haben sich zwei Risiken verwirklicht: zum einen die nicht regelrechte Implantatlage, zum anderen die nicht rechtzeitige Behandlung bei Fehllage.
Nachvollziehbarerweise hätten die Ärzte die korrekte Lage des verwendeten Implantats intra- und postoperativ stets unter Kontrolle halten müssen. Bei Anhaltspunkten für eine falsche Positionierung wäre rasches Handeln angezeigt gewesen. Den Ärzten ist insbesondere der Vorwurf zu machen, dass sie trotz bereits vermuteter instabiler Situation keine sofortige Lösung in Form einer Revisions-OP herbeiführten. Nicht zuletzt aus dieser Verzögerung ergibt sich die erhebliche Regulierungssumme.
Fall 2
Einer 59-jährigen Frau wird im Rahmen eines stationären Krankenhausaufenthalts eine Hüft-TEP implantiert. Postoperativ zeigt sich eine ausgeprägte Beinlängendifferenz nebst Schmerzen und Bewegungsstörungen. Zwei Revisionsoperationen werden notwendig. Die Patientin, die sich mit diesem Verlauf nicht einverstanden erklären will und kann, schaltet die Schlichtungsstelle der zuständigen Ärztekammer ein. Ihr Hauptvorwurf: ein zu großer Prothesenkopf sei eingesetzt worden.
Das Krankenhaus kommt zum Ergebnis, dass der Argumentation der Patientin nichts entgegengehalten werden kann. Bei der Prüfung der Vorwürfe stellt sich heraus, dass die verantwortlichen Ärzte bei einer 28er-Pfanne ein 32er-Inlay verwendet haben, was die beklagten Folgeprobleme wegen deutlicher Beinlängendifferenz auslöste. Hinzu kommt, dass die Prothese nicht ausreichend tief implantiert war.
Die nun angesetzte Revisions-OP bringt aber keine Abhilfe, weil erneut ein 32er-Kopf aufgesetzt wird, ohne die Pfanne abermals zu kontrollieren. Erst bei einer nunmehr dritten (!) OP in einem anderen Krankenhaus wird der Fehler erkannt und revidiert.
Angesichts der Stellungnahme der behandelnden Ärzte kommt der Haftpflichtversicherer des Krankenhauses um eine Zahlung nicht herum. Insgesamt reguliert er gut 38.000 €.
Risikobewertung
Auf die Größe kommt es an – so könnte man das Problem in diesem Fall umschreiben. Tatsächlich handelt es sich aber nicht nur um eine – zweimalige – fahrlässige Verwechslung der Inlay-Größe, sondern auch um schwerwiegende Kontrollfehler, was aus Sicht der Patientin verständlicherweise kaum mehr nachzuvollziehen ist.
Beschwerden und Vorwürfe wegen falscher Prothesengrößen kommen in der Schadenbearbeitung verhältnismäßig häufig vor. Die in diesem Bereich tätigen Ärzte sollten sich bei der Wahl der Prothese bzw. der Prothesenkomponenten des erhöhten Risikopotenzials bewusst sein.
Fall 3
Ein 70-jähriger Patient hat eine Hüft-TEP implantiert bekommen. Postoperativ kommt es zu einigen Problemen im operierten Bereich. Später stellt sich heraus, dass eine Darm-Leckage besteht und sich ein Abszess gebildet hat, der wiederum eine generalisierte Infektion verursacht. Nach drei Wochen stirbt der Patient.
Die Angehörigen bitten die Schlichtungsstelle der Ärztekammer um Überprüfung der zurückliegenden Behandlung. Im Krankenhaus, so ihr Vorwurf, seien nicht alle notwendigen diagnostischen Maßnahmen durchgeführt worden. In erster Linie ist nach Meinung der Angehörigen die Hüfte des Patienten nicht ausreichend untersucht worden, obwohl er über Schmerzen in diesem Bereich geklagt habe. Erst auf ihr Drängen hin habe man sich dazu entschlossen, die voroperierte Hüfte erneut zu öffnen. Dabei sei endlich der Abszess als Ursache der generalisierten Infektion festgestellt worden. Die OP sei jedoch zu spät erfolgt. Nach Ansicht der Angehörigen würde der Patient noch leben, wenn die Ursache der Infektion eher gefunden worden wäre.
Die behandelnden Chirurgen hingegen sind der Ansicht, dass zunächst keine Hinweise auf eine Infektion des Hüftgelenks vorgelegen hätten. Auch hätten sich die Beschwerden in diesem Bereich zeitweise gebessert. Der Entschluss zur TEP-Revision sei als ultima ratio gefasst worden, nachdem sich der Zustand des Patienten weiter verschlechtert habe. In einem Becken-CT seien zuvor nur kleine gluteale Abszesse mit nur fraglichem Kontakt zur Hüft-TEP-Pfanne festgestellt worden. Ein Keimnachweis jedoch sei bei der Revision überraschenderweise nicht gelungen. Insgesamt sehen die Ärzte kein fehlerhaftes Verhalten bei sich selbst.
 Das sieht der Gutachter der Ärztekammer allerdings anders. Man habe nicht in ausreichendem Maße nach dem Infektionsfokus gesucht. Wahrscheinlich sei eine periprothetische Infektion des linken Hüftgelenks Ursache der Sepsis beim Patienten gewesen. Die Prüfung, ob eine solche vorliegt, hätte daher nach Ansicht des Gutachters früher erfolgen müssen. Eine rechtzeitige Therapie – mit Explantation der infizierten Hüftendoprothese, Sanierung des Infektionsherdes und, in einem zweiten Eingriff, Implantation einer neuen Prothese – hätte die Chance auf Heilung auch nach Einschätzung des Gutachters maßgeblich erhöht.
Das sieht der Gutachter der Ärztekammer allerdings anders. Man habe nicht in ausreichendem Maße nach dem Infektionsfokus gesucht. Wahrscheinlich sei eine periprothetische Infektion des linken Hüftgelenks Ursache der Sepsis beim Patienten gewesen. Die Prüfung, ob eine solche vorliegt, hätte daher nach Ansicht des Gutachters früher erfolgen müssen. Eine rechtzeitige Therapie – mit Explantation der infizierten Hüftendoprothese, Sanierung des Infektionsherdes und, in einem zweiten Eingriff, Implantation einer neuen Prothese – hätte die Chance auf Heilung auch nach Einschätzung des Gutachters maßgeblich erhöht.
Die Schlichtungsstelle der Ärztekammer schließt sich dem Gutachten an und erlässt einen für die beteiligten Ärzte negativen Bescheid. In der Summe liege ein grober Behandlungsfehler vor. Die eingetretene Verzögerung sei geeignet gewesen, das Fortschreiten der Sepsis und damit den Tod des Patienten zu diesem Zeitpunkt zu verursachen.
Insgesamt zahlt der Haftpflichtversicherer über 77.000 € an die Angehörigen und die Krankenkasse.
Risikobewertung
Die Sorge um ein Implantat endet nicht mit dem Abschluss der Operation. Auch die Nachsorge – vor allem bei postoperativen Beschwerden – darf nicht vernachlässigt werden und ist mit der entsprechenden Um- und Weitsicht umzusetzen. Dass hierbei auch und besonders eine mögliche Infektion in die Überlegungen mit einbezogen werden muss, zeigt dieser Fall. Hinzu kommt der Zeitfaktor, der hier die entscheidende Rolle spielt.
Der beschriebene ist nicht der einzige Fall in der Schadendatenbank der Ecclesia, der mit einer falschen Behandlung bei Infektion nach Implantation einer Hüft-TEP im Zusammenhang steht. Die Ärzte, Chirurgen und Orthopäden sollten sich der Gefahr einer Infektion bei oder nach einer Hüft-TEP-OP daher stets gewahr sein.
Die haftungsrechtlichen Risiken bei der Implantation einer Hüft-TEP, die wir hier aufgezeigt haben, zählen zu den „Klassikern“, weil sie für einen großen Teil der Schadenfälle im Bereich Hüft-TEP verantwortlich sind:
- die Gefahr einer Infektion,
- die Wahl der falschen Größe der Prothese und
- die falsche Lage der Prothese.
Vorwürfe wegen fehlerhafter Kontrolle des Operationsergebnisses und wegen verzögerter weiterer Diagnostik und Behandlung können im Schadenfall die Folge sein. Wenn Ärzte die Risiken aber im Blick haben und entsprechend (re)agieren, tragen sie entscheidend dazu bei, solche Schadenfälle zu verringern.
Im Folgenden geben wir einen Überblick über sinnvolle schadenpräventive Maßnahmen im Allgemeinen und, auf die beschriebenen Fälle bezogen, im Besonderen.
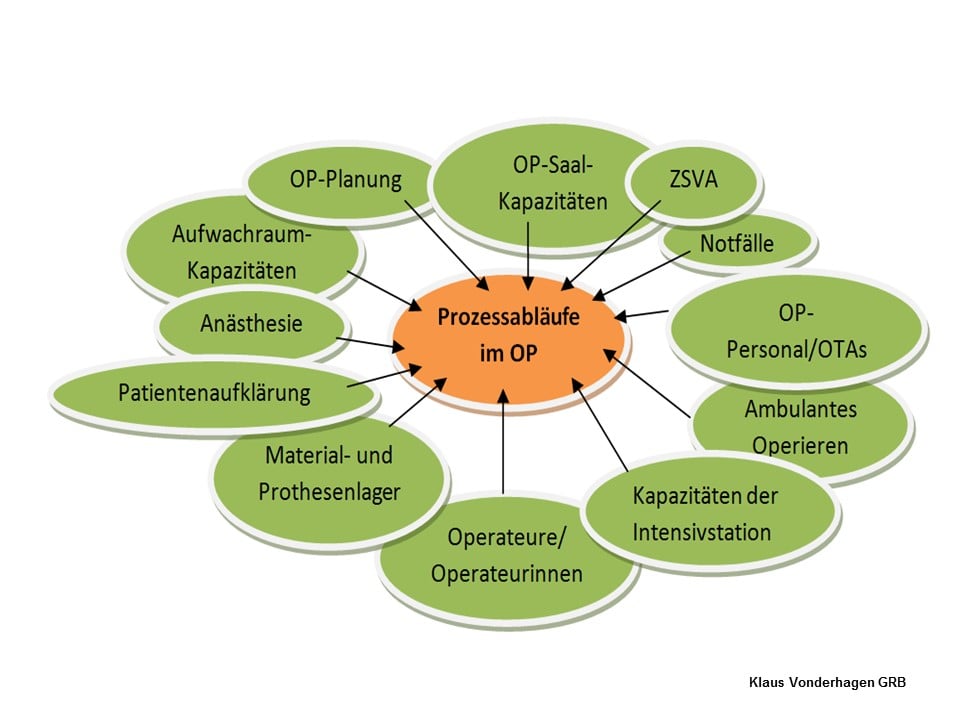
Präoperative Maßnahmen im Sinne eines klinischen Risikomanagements
Kommt der Patient ins Krankenhaus/in die Klinik, um sich einem elektiven operativen Eingriff wie z. B. der Implantation einer Hüftprothese zu unterziehen, geschieht dies in der Regel nicht von heute auf morgen, sondern nach Terminvereinbarung. Daher sind hier die präoperativen Maßnahmen gut plan- und rechtzeitig machbar.
In der Regel beginnt die Kontrolle, ob die notwendigen Prozessschritte eingehalten worden sind, in der Indikationssprechstunde. Im Folgenden listen wir die einzelnen Schritte/Maßnahmen auf, die zum jeweiligen Zeitpunkt erledigt sein müssen bzw. zu erledigen sind.
Präoperativ vor Aufnahme des Patienten bei der Indikationssprechstunde
- Die wesentlichen Patientendaten sind vorhanden.
- Patientenbezogenes Bildmaterial liegt vor und es ist aussagekräftig.
- Die Therapieplanung steht fest und wurde im Rahmen einer OP-Terminanfrage (z. B. Aufnahme in den „OP-Monatsplan“) mit dem OP-Management besprochen.
- Bei entsprechendem Anlass wurde ein MRSA-Screening beim Patienten und, wenn notwendig, eine Keimreduzierung auf der Haut und Nasenschleimhaut vorgenommen (fünf Tage vor OP mit entsprechenden Maßnahmen festgelegt).
- Das System/die Prothese wurde für den Patienten ausgesucht, bestellt und reserviert (sollte 48 Stunden vor OP-Beginn zur Verfügung stehen).
- Der Patient wurde der zuständigen Physiotherapie zur Erhebung einer Anamnese, Ermittlung des Gangbilds und Anpassung der notwendigen postoperativen Hilfsmittel (z. B. Unterarmgehstützen, Sitzerhöhung) vorgestellt.
- Patientenaufklärungen (Operateur und Anästhesie) wurden rechtzeitig durchgeführt und von den aufklärenden Ärzten sowie dem Patienten unterschrieben.
Nach Aufnahme in das Krankenhaus/die Klinik, in der Regel am Vortag zur OP
- Der Patient erhält ein Identifikationsarmband mit seinen Daten (Name, Vorname, Geburtsdatum).
- Das OP-Management bestätigt, dass das System/die Prothese für den Patienten zur Verfügung steht.
- Der Operateur hat das Operationsgebiet gekennzeichnet.
- Die entsprechende Operation ist nach dem OP-Planungsgespräch mit dem Patienten im OP-Plan eingetragen worden und hat dort eine feste Position.
- Besonderheiten beim Patienten (z. B. Herzschrittmacher, Begleiterkrankungen, weitere Implantate, Metallteile im oder am Körper, eine Infektion etc.) sind im OP-Plan vermerkt.
Am OP-Tag
- Für den Patienten wurde die OP-Checkliste angelegt.
- Der präoperative Teil wurde im stationären Bereich abgearbeitet.
Im OP
Nach Übergabe des Patienten an der OP-Schleuse an die Mitarbeiter im OP übernimmt eine vom Krankenhaus/von der Klinik speziell aufgestellte OP-Checkliste die Abfrage der definierten Sicherheitskriterien für die jeweiligen Zeitabschnitte:
- Sign In
- Team Time Out
- Sign Out
Bezogen auf die Risiken aus den drei o. g. Fallbeschreibungen könnten die Präventionsmaßnahmen in der OP-Checkliste beispielsweise wie folgt aussehen:
- 30 Minuten vor Hautschnitt (Sign In-Phase): Als mögliche Prävention zur Vermeidung von postoperativen Infektionen erfolgt – nur nach Anordnung des Operateurs – eine Antibiotikagabe als „single shot“.
- vor der sterilen Übergabe der Prothese an die OP-Schwester: Der OP-Springer zeigt dem Operateur die Prothese (Hüftkopf und Hüftpfanne) einschließlich Größenangaben unter Einsatz der „Call-Recall-Methode“.
- nach dem Einsetzen der Prothese: Ihr Sitz wird mit einer Röntgenkontrolle (z. B. intraoperative 3D-Röntgenvisualisierung) im OP überprüft und mittels Bild dokumentiert.
|
vor Anästhesiebeginn in der Einleitung
„Sign In“
|
vor der Hautinzision
„Time Out“
|
Vor Verlassen des OP
„Sign Out“
|
Fazit
Eine Prozesskette wie beschrieben aufzubauen, erhöht die Patientensicherheit und hilft, Schäden an Patienten zu vermeiden. Um dies zu erreichen, gibt die Krankenhaus-/die Klinikleitung zunächst die Struktur für ein klinisches Risikomanagement vor. Im nächsten Schritt werden die bestehenden Prozessabläufe auf mögliche Risiken hin überprüft. Aus den so identifizierten Risiken können dann Präventionsmaßnahmen abgeleitet werden, die schließlich in den Prozessablauf eingebettet und verbindlich zur Anwendung gebracht werden.