Allgemeines
Weichteilsarkome sind bösartige Tumoren mesenchymalen Ursprungs, die insgesamt nur ca. 1 Prozent aller malignen Erkrankungen ausmachen [1]. Wie alle bösartigen Tumoren zeichnen sie sich durch ein unterschiedlich aggressives, invasives Wachstum und eine potenzielle Metastasierungsfähigkeit (überwiegend pulmonal) aus. Die Inzidenz wird für Europa und Nordamerika derzeit mit ein bis zwei Neuerkrankungen pro 100.000 Einwohner und Jahr angegeben [2, 3]. Sie sind somit häufiger als Tumoren des Knochens oder Knorpels, jedoch deutlich seltener als Kolon- oder Bronchialkarzinome [4]. Leider existieren in Deutschland keine epidemiologischen Daten zu Weichteilsarkomen. So sammelt z. B. das Robert Koch-Institut keine Daten zu Weichteilsarkomen (Zuordnung ICD C45-49).
Lokalisation
Weichteilsarkome treten an nahezu allen Lokalisationen auf, wobei sich ihre Verteilung auf die einzelnen Körperregionen proportional zum Anteil der Weichgewebsmasse der jeweiligen Köperregionen verhält [5]. In 50 Prozent aller Fälle gehen Weichteilsarkome aus dem Binde- und Stützgewebe der unteren Extremität hervor, während 10 Prozent an der oberen Extremität entstehen. Ein Drittel der Weichteilsarkome findet sich im Rumpf mit Prädilektion im Retroperitoneum, wohingegen der Kopf-Hals-Bereich mit 10 Prozent seltener betroffen ist. Aktuelle Daten belegen eindeutig, dass Tumorgröße und -Lokalisation prognostisch relevant sind [5].
Klinik
Weichteilsarkome an den Extremitäten treten zumeist als tast- und sichtbare schmerzlose Schwellungen in Erscheinung. Schlechter differenzierte Weichteilsarkome können zudem eine derbe Konsistenz und eine geringere Verschieblichkeit aufweisen. In manchen Fällen kann durch die Gewebeverdrängung eine verstärkte lokale Venenzeichnung an der darüberliegenden Haut festgestellt werden. Umgebungsreaktionen wie Rötungen und Erwärmungen treten allerdings erst sehr spät bei exulzerierenden Verläufen auf. Das Auftreten von Schmerzen ist in erster Linie abhängig von der Lokalisation des Tumors, jedoch in der Regel Folge einer Infiltration oder Verdrängung von Nerven und ist somit meistens als Spätsymptom anzusehen. Es fehlen weitestgehend Krankheitszeichen im Sinne einer B-Symptomatik wie Gewichtsverlust, Fieber oder Nachtschweiß. Eine Inappetenz wird ebenfalls fast nie angegeben. Im Gegensatz zu Karzinomerkrankungen treten paraneoplastische Syndrome nur in den seltensten Fällen auf, was zur Bagatellisierung des Befundes führen kann. Nicht selten ist die Erkrankung bei Diagnosestellung lokal weit fortgeschritten oder hat sogar Metastasen gebildet.
Metastasierung
Ungefähr 30 Prozent aller Betroffenen entwickeln im Krankheitsverlauf hämatogene, zumeist pulmonale Fernmetastasen [6, 7]. Lymphknotenmetastasen hingegen sind insgesamt selten und treten nur bei einigen wenigen Subtypen wie Rhabdomyosarkomen, Klarzellsarkomen und epitheloidzelligen Sarkomen auf [8, 9]. Das mediane Überleben nach diagnostizierter Fernmetastasierung liegt bei weniger als 15 Monaten [6].
Heterogenität
Weichteilsarkome zeichnen sich mit über 50 bekannten Subtypen durch eine ausgeprägte Heterogenität aus, die sich nicht nur in dem erreichten Phänotyp, sondern auch in der klinischen Manifestation und Prognose zeigt. [10, 11]. Von daher ist die richtige histopathologische Diagnose von essenzieller Wichtigkeit. Die histologische Sicherung ist allerdings weiterhin schwer und erfordert molekularpathologische Techniken. Außerhalb der großen Zentren sollte die Diagnose immer durch einen Referenzpathologen abgesichert werden [12].
Am häufigsten treten im Erwachsenenalter das Liposarkom, das NOS (not otherwise specified sarcoma) und das Leiomyosarkom auf [3, 4]. Gerade aufgrund der Heterogenität und der geringen Fallzahlen ist es schwierig eine sichere prozentuale Angabe zu den Häufigkeiten der einzelnen Subentitäten zu geben, da diese je nach Land und Referenzzentrum deutlichen Schwankungen unterliegen. Wenn man alle Entitäten berücksichtigt, dann liegt der Altersgipfel für eine Neuerkrankung zwischen 60 und 65 Jahren [4]. Natürlich muss hierbei bedacht werden, dass die histologischen Subtypen altersbezogene Häufigkeitsgipfel zeigen. So treten in der Altersgruppe der über 60-jährigen kaum Rhabdomyosarkome auf, während im Kindes- und Jugendalter diese Entität eines der häufigsten soliden Tumorerkrankungen überhaupt darstellt. [13, 14].
Diagnostik
Bei unklaren Weichteilschwellungen an den Extremitäten sowie im Brust- und Bauchwandbereich ist die Magnetresonanztomographie unter intravenöser Gadolinium-Kontrastmittelgabe das am geeignetste bildgebende Verfahren [15]. Mit ihrer Hilfe kann die exakte Tumorausdehnung samt Lagebeziehung zu Nerven, Gefäßen und Knochen genau beurteilt werden. Weiterhin kann eine erste Abschätzung hinsichtlich der Differenzierung getroffen werden. Oftmals zeigen sich bei großen, schlecht-differenzierten Läsionen bereits intratumorale Nekrosen. Nichtsdestotrotz ist für die weitere Therapieplanung eine histopathologische Untersuchung der suspekten Weichteiltumoren erforderlich [16]. Differentialdiagnostisch sollten Lymphome, Weichteilmetastasen von Karzinomen und eventuelle entzündliche Prozesse ausgeschlossen werden, da sie therapeutisch ein komplett anderes Vorgehen erfordern. Die Probenentnahme erfolgt an den Extremitäten und den Körperwänden in der Regel mittels Ultraschall- oder CT-gesteuerter Stanzbiopsie. Bei kleineren Läsionen, die mittels Stanze nur unsicher erfasst werden können, kann in unkomplizierten Fällen und oberflächlicher Lage auch eine Exzisionbiopsie erwogen werden. Ansonsten sollte, wenn immer möglich, vor definitiver operativer Versorgung die Tumorentität samt Malignitätsgrad bekannt sein und eine Behandlungsstrategie im Tumorboard erarbeitet sein. Bei G2- und G3-Weichteilsarkomen sollte präoperativ eine CT-Untersuchung des Thorax zur Abklärung eventueller Fernmetastasen durchgeführt werden [17].
Therapie
Therapie bei lokalisiertem Befall
Die relative Seltenheit der einzelnen Entitäten setzt eine außerordentliche Erfahrung in der Therapie der Weichgewebssarkome voraus. Das therapeutische Konzept ist daher interdisziplinär und multimodal, sowohl die operative Therapie als auch die Strahlen- und die Chemotherapie sind von Bedeutung und werden interdisziplinär im Tumorboard festgelegt. Die Therapie der Wahl bei Weichteilsarkomen mit lokalisiertem Befall besteht weiterhin in der kompletten chirurgischen Entfernung im Gesunden. Das Ziel der operativen Therapie ist die Erlangung tumorfreier Resektionsränder im Sinne einer R0-Resektion. Dies bedeutet, der Tumor wird en bloc in No-Touch-Technik operiert. Frühere Angaben zu Sicherheitsabständen (5 cm zur Seite, 2 cm zur Tiefe) entsprechen Expertenvorstellungen und sind nie mit Daten hinterlegt worden.
Das chirurgische Behandlungskonzept samt Resektionsumfang muss der Lokalisation angepasst und bei jedem Patienten individuell bestimmt werden. Inwieweit der Sicherheitsabstand im Gesunden einen prognostischen Einfluss hat, wird derzeit noch kontrovers diskutiert. Die bisher durchgeführten retrospektiven Analysen, die den Einfluss der Resektionsabstände untersucht haben, konnten allerdings keinen prognostischen Unterschied zwischen knappen und weiten Sicherheitsabständen ausmachen [18-20]. Eine eigene retrospektive Analyse von 700 Patienten untermauert diese Angaben (in Präparation). Prospektiv randomisierte Daten fehlen.
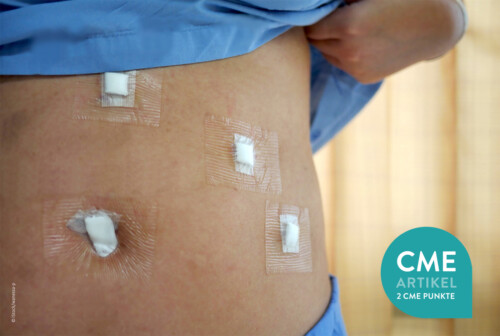
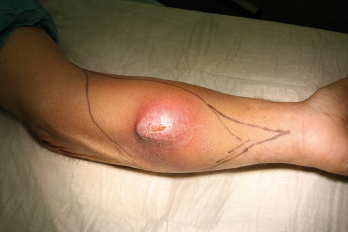
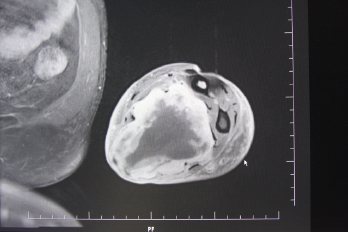
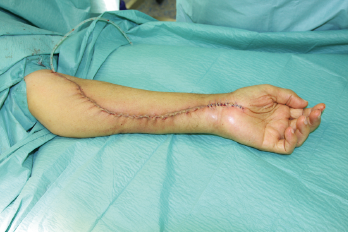
In der eigenen Klinik wird seit Beginn der 90er Jahre mit nunmehr über 3.500 operierten Patienten die Taktik der R0-Resektion unter Toleranz auch geringerer Sicherheitsabstände durchgeführt (Abb. 1-5). Die vergleichende Analyse der onkologisch relevanten Parameter scheint diese Vorgehensweise zu bestätigen.
|
|
|
|
|
 |
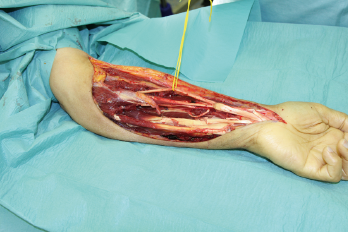
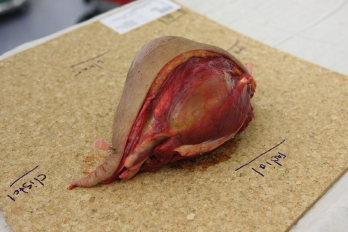
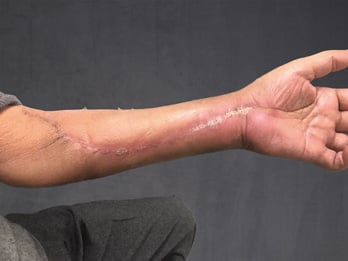
Der Stellenwert der adjuvanten Chemotherapie mit den Erstlinienchemotherapeutika Doxorubicin und Ifosfamid ist bei primär resektablen Weichteilsarkomen weiterhin umstritten. Die bisher größte zur adjuvanten Chemotherapie durchgeführte Meta-Analyse konnte nur einen marginalen Einfluss auf das krankheitsspezifische Gesamt-Überleben nach zehn Jahren feststellen [30]. Eine prospektiv, randomisierte Studie der European Organisation for Research and Treatment of Cancer (EORTC 62931) konnte ebenfalls keinen Überlebensvorteil durch eine adjuvante Chemotherapie feststellen [31]. Letztendlich wurde empfohlen in zukünftigen Studien zur adjuvanten Chemotherapie nur Patienten mit großen G3-Extremitätensarkomen einzuschließen, da diese Hochrisiko-Patienten unter Umständen von einer adjuvanten Chemotherapie profitieren könnten, wobei die Ergebnisse dieser Studien abzuwarten sei.
Interessant erscheint die Tatsache, dass weiterhin nur vier Substanzen für die Behandlung zugelassen sind (Doxorubicin gehört nicht dazu), aber sicher zehn unterschiedliche Chemotherapeutika Anwendung finden.
Die neoadjuvante Chemotherapie konnte sich aufgrund fehlendem signifikantem Überlebensvorteil nicht durchsetzen und sollte daher auf Studien beschränkt bleiben.
Therapie bei metastasiertem Krankheitsverlauf
Bei solitären, pulmonalen Metastasen wird eine komplette chirurgische Resektion des Primärtumors und der Metastase angestrebt sowie eine adjuvante oder neoadjuvante Chemotherapie durchgeführt, um die nunmehr systemischen Erkrankung zu behandeln [6]. Bei disseminierten Metastasen sind die Therapieoptionen jedoch palliativer Natur und zumeist auf die Chemotherapie beschränkt. Etabliert hat sich hierbei seit Jahrzehnten das Anthrazyklin Doxorubicin, welches als Monotherapeutikum bei metastasierten Krankheitsverläufen Ansprechraten von 20 bis 30 Prozent vorweist [14]. Durch die Kombination mit dem Oxazaphosphorin Ifosfamid können zwar leicht verbesserte Ansprechraten erzielt werden, die jedoch nicht mit einem Überlebensvorteil einhergehen [32]. Bei Patienten mit metastasierten Leiomyosarkomen oder Liposarkomen konnte zuletzt im Rahmen von Phase-3-Studien eine gewisse Wirkung für Trabectedin und Eribulin ausgemacht werden, sodass ihr Einsatz bei diesen Subtypen empfohlen wird, wenn Doxorubicin nicht ansprechen sollte [33, 34]. Bei Patienten, die an disseminierten nicht-lipomatösen Weichteilsarkomen leiden, kann nach frustraner Doxorubicin-Behandlung eine Therapie mit Pazopanib erwogen werden [35].
Nachsorge
Auch hier gibt es aufgrund fehlender Evidenz keine einheitliche Aussage. In der eigenen Klinik führen wir bei Patienten mit G2- und G3-Sarkomen für die ersten zwei Jahre alle drei Monate eine klinische Verlaufskontrolle samt MRT-Untersuchung des Lokalbefundes und Röntgenuntersuchung des Thorax in zwei Ebenen durch. Nach zwei Jahren wird das Untersuchungsintervall auf sechs Monate verlängert. Je nach Verlauf werden die Kontrollen nach insgesamt fünf Jahren fortgesetzt oder gar beendet. Inwieweit eine weitere Untersuchung über fünf Jahre hinaus sinnvoll ist, ist noch nicht geklärt [22]. Bei Patienten mit G1-Sarkomen führen wir die Kontrollen in den ersten fünf Jahren halbjährlich durch. Ein ähnliches Schema wird aktuell auch von der ESMO empfohlen, wobei der Nutzen einer routinemäßig durchgeführten MRT-Nachsorgeuntersuchung uneinheitlich bewertet wird [22, 23].
Fazit
Insgesamt sollte die Behandlung der Weichgewebssarkome aufgrund der Seltenheit, Heterogenität und unklaren Datenlage dafür spezialisierten Zentren vorbehalten sein.
Nur so kann die aktuell hohe Rate falscher Primärbehandlungen gesenkt werden.
Aufgrund der breiten Lokalisationsverteilung muss das Weichgewebssarkom als „Querschnittsmalignom“ bezeichnet werden, dem eine klare Fachdisziplin-Zuordnung fehlt. Damit verbleibt die Therapie mit einer hohen Interdisziplinarität behaftet.
Zur Verbesserung der Behandlungsqualität erfolgt aktuell unter Federführung der interdisziplinären Arbeitsgruppe Weichgewebssarkome (IAWS) der Deutschen Krebsgesellschaft (DKG) die Erstellung einer S3-Leitlinie entsprechend dem Leitlinienprogramm Onkologie der DKG, der Deutschen Krebshilfe (DKH) und der AWMF unter Zugrundelegung der durch diese Gesellschaften festgelegten Qualitätsindikatoren. Damit wird erstmalig eine S3-Leitlinie zu einer Gruppe der seltenen Tumoren erarbeitet.
Weiterhin definiert die IAWS in Kooperation mit Onkozert aktuell Qualitätsindikatoren, die für die Etablierung von Zentrumszertifizierungen entsprechend der onkologischen Zentren (OZ) als Modul für die Behandlung der Weichgewebssarkome zugrunde gelegt werden.
Literatur via passion_chirurgie@bdc.de


Lehnhardt M. / Hirsch T. / Behr B. / Harati K. CME-Artikel: Weichteilsarkome – Verbesserung der Behandlung durch Leitlinienbildung und Zentrumszertifizierung. Passion Chirurgie. 2017 Juni; 7(06): Artikel 03_04.